Die spinale Muskelatrophie wird durch eine Mutation im Survival-of-Motor-Neuron-1(SMN1)-Gen verursacht. Die Mutation des Gens führt zu geringerer Produktion des SMN-Proteins, was für die Funktionalität der motorischen Nervenzellen erforderlich ist.
Auf den verwendeten Bilder sind keine echten Patienten dargestellt; sie dienen nur zur Veranschaulichung.
Was ist die Ursache der SMA?
Ursache der spinalen Muskelatrophie ist eine Mutation im Survival-of-Motor-Neuron-1 (SMN1)-Gen. Dieses Gen ist für die Bildung des Survival-Motor-Neuron (SMN)-Proteins verantwortlich, das für die normale Funktion der motorischen Nervenzellen erforderlich ist. Bei Personen mit spinaler Muskelatrophie sind beide Kopien des SMN1-Gens mutiert, was zu einer verminderten Produktion des SMN-Proteins führt. Ohne eine angemessene Menge an SMN-Protein gehen die motorischen Nervenzellen im Rückenmark zugrunde, sodass die Muskeln keine Signale vom Gehirn mehr empfangen können.4,5
Der Untergang von motorischen Nervenzellen führt zur stetigen Abnahme von Muskelmasse und
Muskelstärke (Atrophie)
Adaptiert nach Arkblad et al, 2009; Jedrzejowska et al, 2010; Prior, 2010; Sugarman et al, 2012; Ogino et al, 2004.14,18-22
Adaptiert nach Norwood et al, 2009; Jones et al, 2015.6,23
WAS MUSS ICH ÜBER SMN2 WISSEN
Sowohl gesunde Menschen als auch Personen mit spinaler Muskelatrophie haben mindestens ein „Ersatzgen“, das als SMN2 bezeichnet wird. Das SMN2-Gen hat eine ähnliche Struktur wie SMN1, doch nur ein kleiner Teil (10 %) des mithilfe des SMN2-Gens produzierten SMN-Proteins ist voll funktionsfähig. Diese geringe Menge an SMN-Protein reicht nicht aus, um die motorischen Nervenzellen im ZNS am Leben zu halten.12,13
Die Anzahl der SMN2-Gene kann unterschiedlich sein. Eine höhere Zahl an SMN2-Kopien ist vermutlich mit weniger schweren Symptomen der spinalen Muskelatrophie verbunden.4,13,14 Trotzdem zeigt die Erkrankung ein breites Spektrum an Symptomen. Es ist kaum möglich, den Schweregrad der Erkrankung allein anhand der Anzahl der SMN2-Kopien vorherzusagen. Deswegen empfehlen Experten, dass Therapieentscheidungen anhand der erreichten Fähigkeiten des Betroffenen und nicht nur anhand der Anzahl an SMN2-Kopien getroffen werden.2
Die Entdeckung des SMN2 Ersatzgens eröffnete eine einzigartige Möglichkeit zur Entwicklung von Therapien, die das SMN2-Gen dahingehend beeinflussen, dass eine höhere Menge an SMN-Protein produziert wird.
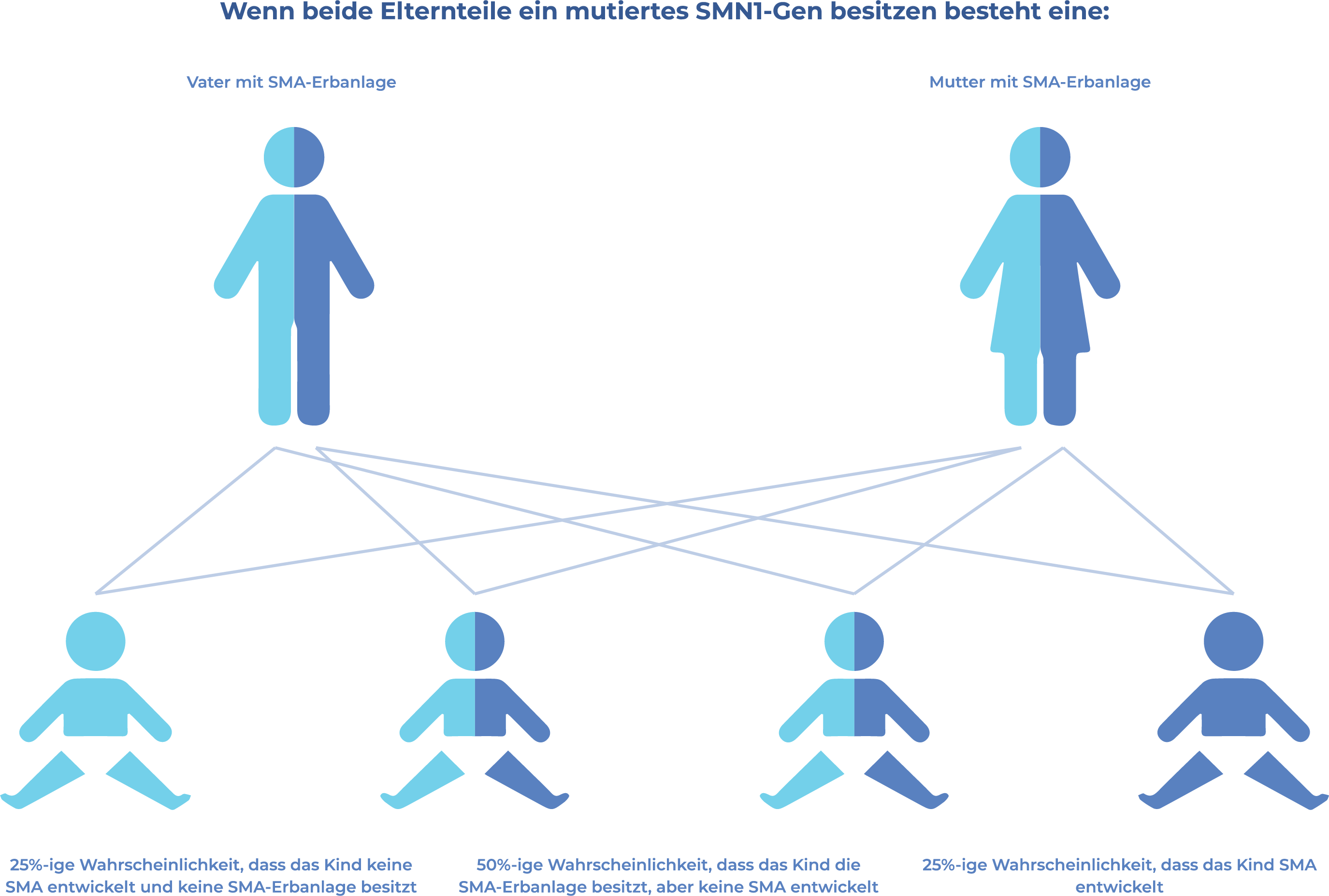
WIE WIRD SMA VERERBT?
Die spinale Muskelatrophie ist eine autosomal rezessive Erkrankung, was bedeutet, dass ein Erkrankungsrisiko nur dann besteht, wenn das Kind zweier SMA-Träger ein mutiertes SMN1 -Gen von jedem Elternteil erbt. Wenn ein Kind nur 1 mutiertes SMN1 -Gen erbt, wird es als „Überträger" betrachtet, entwickelt für gewöhnlich jedoch keine Symptome der spinalen Muskelatrophie.16
Wenn es in Ihrer Familie Fälle von spinaler Muskelatrophie gab, ist die Wahrscheinlichkeit, dass Sie Träger der SMA-Erbanlage sind, höher als der Durchschnitt. Bei der Familienplanung sollten Sie deswegen Ihren Arzt um Rat fragen, um zu erfahren, welche Mutationen in Ihrer Familie vermehrt auftreten. Sobald die in Ihrer Familie vorhandenen Mutationen bekannt sind, kann eine für Sie geeignete Untersuchung durchgeführt werden:
Wenn es sich bei den Mutationen in Ihrer Familie um SMN1-Deletionen handelt, kann eine Bestimmung der Anzahl der Genkopien für Sie angemessen sein.
Wenn die Mutationen in Ihrer Familie geringere Genveränderungen umfassen, werden Ihr Arzt und das Labor entscheiden, ob Untersuchungen zum Nachweis spezifischer Veränderungen möglich sind.
Wenn Ihnen keine Informationen über die Mutationen in Ihrer Familie zur Verfügung stehen, können Sie trotzdem die Anzahl an Genkopien analysieren lassen. Die Wahrscheinlichkeit, dass Sie Träger der SMA-Erbanlage sind, wird (vor den Untersuchungen) anhand Ihrer Familiengeschichte ermittelt. Wenn Ihre Ergebnisse normal sind, ist die Wahrscheinlichkeit, dass Sie Träger der SMA-Erbanlage sind, geringer.