Ein Land oder eine Region wählen


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)
-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)

-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)

-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
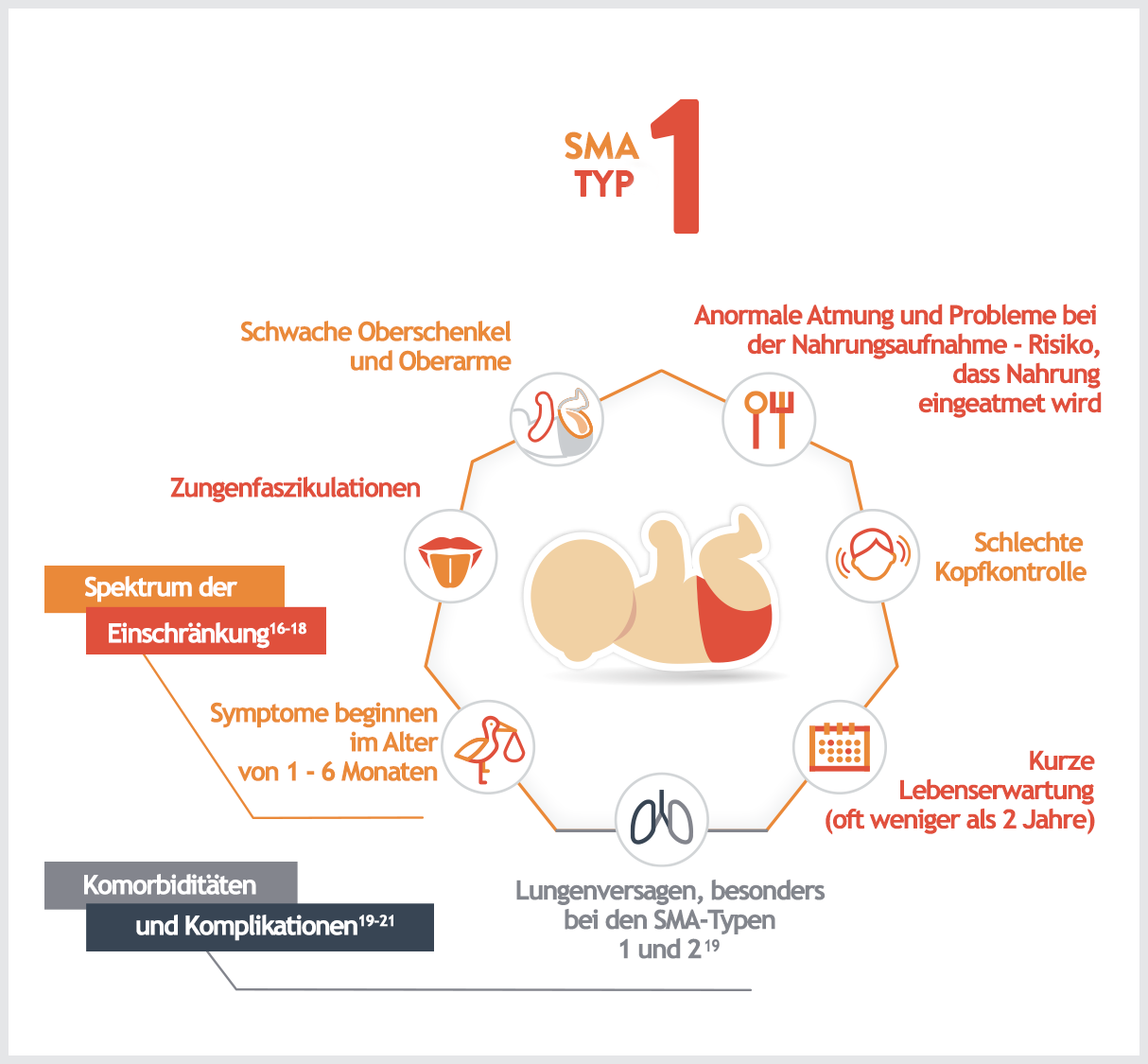
Merkmale:1,4,5,7
- Schlechte Kopfkontrolle
- Schwaches Husten
- Kraftloses Schreien
- Fortschreitende Schwäche der Kau- und Schluckmuskulatur
- Schwacher Muskeltonus
- „Froschschenkelhaltung“ im Liegen
- Ausgeprägte Muskelschwäche auf beiden Seiten
- Fortschreitende Schwäche der Atemmuskulatur (Zwischenrippenmuskeln), was zu einer „glockenförmigen“ Brust und einer Schaukelatmung führt
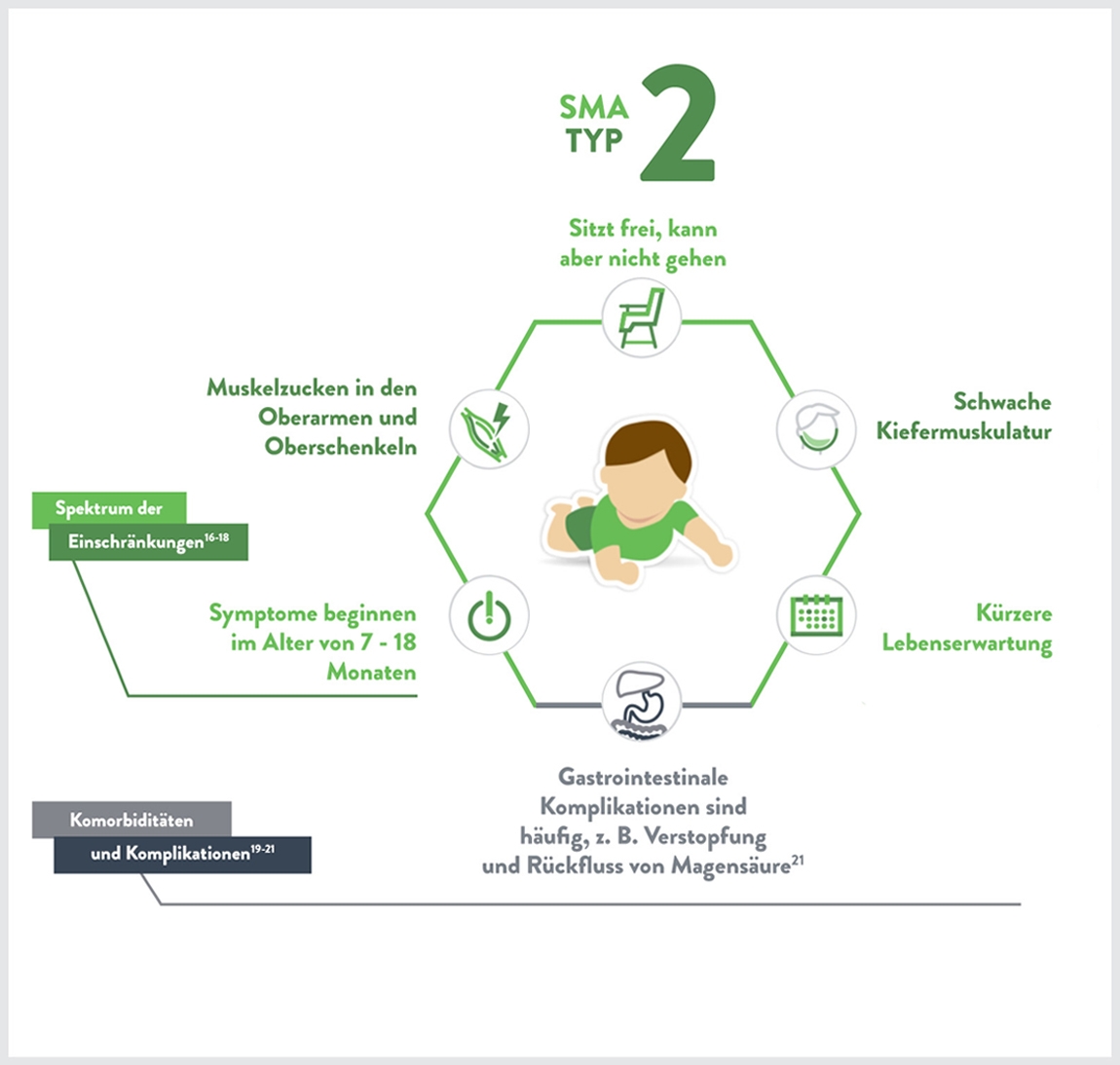
Merkmale:1,5
- Muskelschwäche
- Probleme beim Schlucken, Husten und Atmen können auftreten, sind aber seltener
- Schmerzen in den Muskeln und motorische Einschränkungen (z.B. Kontrakturen)
- Kinder können Wirbelsäulenprobleme wie Skoliose (Krümmung der Wirbelsäule) entwickeln, sodass ein Korsett oder eine Operation erforderlich werden kann
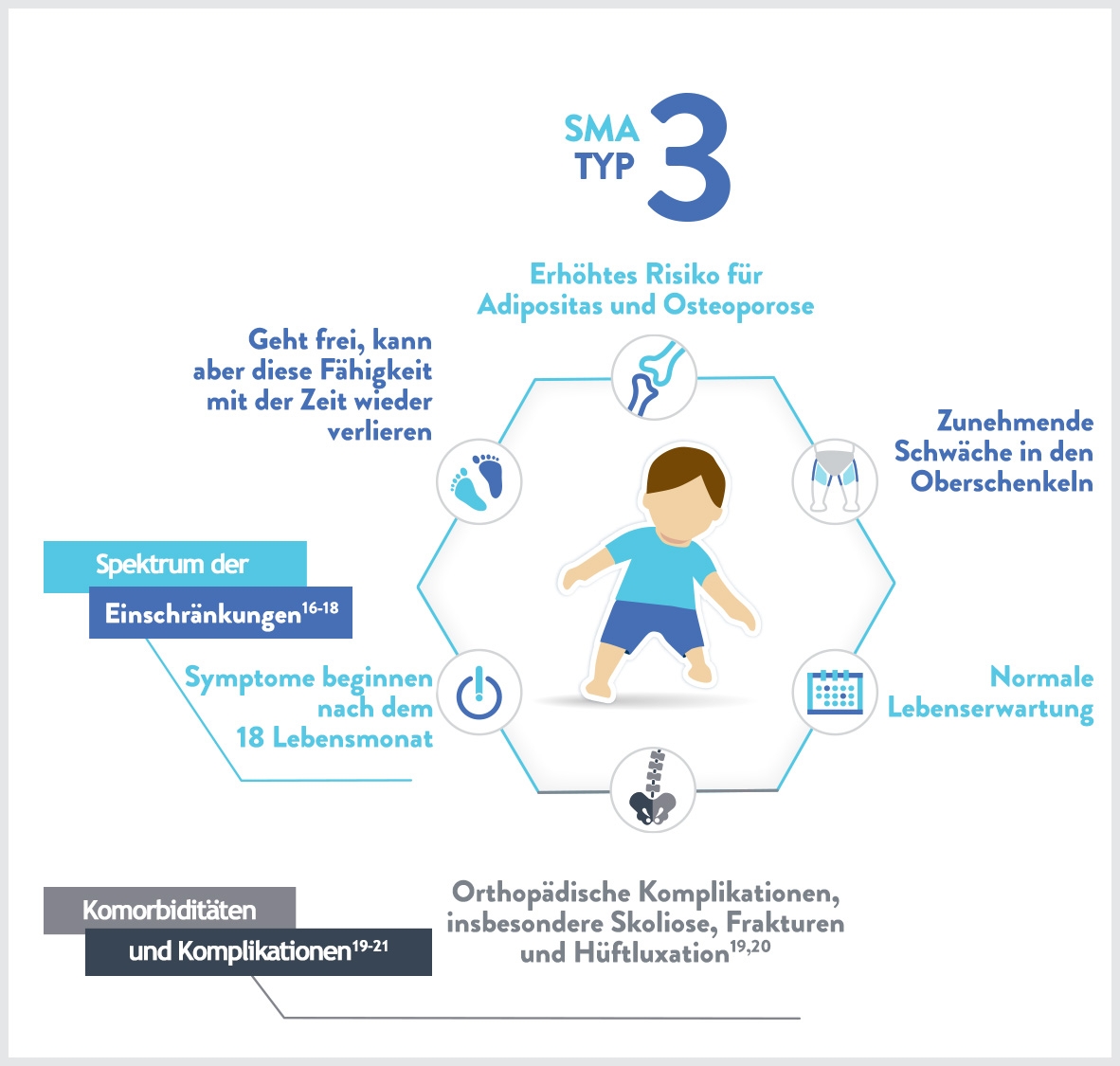
Merkmale:1,5
- Skoliose
- Probleme beim Kauen und Schlucken
- Die Beinmuskeln sind meist stärker betroffen als die Armmuskeln
- Schmerzen in den Muskeln
- Symptome einer Überbeanspruchung der Gelenke
Referenzen
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008; 371 (9630):2120-2133.
10. Iannaccone ST. Modern management of spinal muscular atrophy. J Child Neurol. 2007;22(8):974-978.
20. Prior TW. Spinal muscular atrophy: a time for screening. Curr Opin Pediatr. 2010;22(6):696-702.
25. CureSMA SMArt Moves Checklist for Motor Delays (7-12 months) Website. Accessed August 16, 2023.